Т лимфома кожи
Лимфома кожи
Группа злокачественных опухолей, образующихся в результате бесконтрольного деления (пролиферации) в ней особой группы клеток «белой» крови (лимфоцитов). В зависимости от вида размножающихся лимфоцитов различают лимфомы Т-клеточные (составляют 65-70% случаев) и В-клеточные (20-25% случаев), а также неклассифицируемые лимфомы (10% случаев).
Факторы риска. Развитие лимфомы кожи связано с мутацией Т- или В-лимфоцитов, которая приводит к их бесконтрольному размножению и миграции в кожу. Мужчины страдают от этого заболевания в два раза чаще, чем женщины. Люди среднего и старшего возраста заболевают чаще, чем представители молодого поколения. Случаи развития лимфомы кожи у детей носят единичный характер.
Предполагают, что возникновение злокачественного клона лимфоцитов может быть спровоцировано постоянной антигенной стимуляцией на фоне нарушенной иммунной защиты организма.
Провоцирующую роль отводят таким факторам, как:
генетическая предрасположенность, отягощенный анамнез;
вирусные инфекции – ретровирусы, цитомегаловирусы, простой герпеса 8 типа, вирус Эпштейна—Барра.
действие различных химических веществ и канцерогенов, применяющихся в сельском хозяйстве, химической промышленности, строительстве;
воздействие солнечной радиации (инсоляция);
последствия от приёма некоторых лекарственных средств.
Виды и типы. Различают первичную лимфому (заболевание начинается с поражения дермы) и вторичную (в результате миграции лимфоцитов из лимфоидного органа, в котором происходит их размножение: костный мозг, вилочковая железа, селезенка, лимфоузлы и т.д.).
По степени злокачественности выделяют лимфомы I, II и III степени.
По клиническим проявлениям, выделяют узелковую, бляшечную и эритродермическую формы лимфомы.
Клинические проявления. Характеризуются полиморфизмом сыпи (т.е. имеются различные элементы сыпи — пятна, бляшки, узлы), которые различаются по степени выраженности зуда и увеличением периферических лимфатических узлов.
Узелковая форма Т-клеточной лимфомы кожи I степени — характеризуется мелкими плоскими узелками размером с просяное зерно. Узелки имеют сиреневый или желтоватый цвет, располагаются группами и склонны к спонтанному регрессу. При более злокачественном течении узелки увеличиваются, приобретают вишневый цвет и утрачивают тенденцию к группировке.
Редко встречается мелкоузелковая форма, при которой фолликулярные узелки сливаются в бляшки с напоминающим псориаз поверхностным шелушением. На этом фоне появляются крупные узелки, которые затем подвергаются некрозу.
Бляшечная форма I степени — представлена нерезко отграниченными бляшками желтоватого цвета. Размер бляшек может быть больше ладони. Они постепенно разрешаются с образованием участков атрофии и гиперпигментации.
Бляшечная форма II степени (грибовидный микоз Алибера) — встречается в 26% случаев всех лимфом кожи. Для нее характерно стадиальное развитие. Вначале появляются шелушащиеся ярко-розовые пятна и другие элементы (эритематозная стадия). Затем на месте пятен формируются застойно-красные бляшки часто с мокнущей поверхностью и периферическим ростом (бляшечная стадия). В опухолевой стадии бляшки сменяются плоскими узлами размером до апельсина с некрозом в центре образования.
Эритродермическая форма I степени (синдром пре-Сезари) — часто развивается на фоне длительно, в течение 10-15 лет, существующей экземы или нейродермита. Кожа покрасневшая и отечная, покрыта крупными пластинками белых чешуек. Наблюдается генерализованное увеличение лимфоузлов, дистрофия ногтей, выпадение волос, лихорадка и мучительный зуд.
Через несколько лет процесс переходит в эритродермическую форму II степени (синдром Сезари), характеризующуюся выраженной инфильтрацией, шелушением и сухостью кожи – форма опасная развитием кахексии (истощение).
Первоначальная симптоматика может проявляться общими симптомами наличия онкологического заболевания:
- общей слабостью и быстрой утомляемостью организма;
- ничем не обоснованной апатией;
- повышенной тревожностью и низкой стрессоустойчивостью;
- значительным повышением температуры тела (до 38,0*С);
- ярко выраженной ночной потливостью;
- динамичной и значительной потерей веса;
- нарушением нормального пищеварения.
1. Лабораторыные методы диагностики – клинический анализ крови. При Т-клеточной лимфоме выявляются лейкопения и моноцитоз, при синдроме Сезари лейкоцитоз (до 30000-200000), нейтрофилез, повышение количества эозинофилов.
2. Гистологическое и цитологическое исследование материала, взятого путем биопсии элементов пораженной кожи, а при необходимости – лимфатических узлов . Биопсия позволяет дифференцировать Т- и В- клеточную лимфому кожи, а также определить степень ее злокачественности.
3. При вовлечении в процесс внутренних органов проводят их исследование: УЗИ брюшной полости , рентгенографию легких , КТ орагнов грудной и брюшной полостей, забрюшинного пространства.
При ранней диагностике вышеуказанных методов обследования может быть недостаточно, поэтому применяют:
ИГХ (иммуногистохимический метод) — метод морфологической диагностики, в основе которого лежит визуализация и оценка с помощью микроскопа результатов реакции антиген-антитело в срезах биопсированной ткани;
ПЦР – метод определения клональности с помощью полимеразной цепной реакции. Позволяет определить наличие возбудителя заболевания, даже если в пробе присутствует всего несколько молекул ДНК возбудителя.
Лечение. Основным методом является полихимиотерапия – использование лекарственных средств группы цитостатики, группы кортикостероидов, и интерфероны. В лечении отдельных пятен, бляшек и единичных опухолей применяют:
ПУВА-терапию – метод лечения, который включает использование фотоактивного вещества (псоралены — класс фурокумаринов) совместно с облучением кожи длинноволновым ультрафиолетовым излучением;
Фототерапию . Метод заключается в приёме определённого количества фотосенсибилизаторов, которые, скопившись в опухоли, помогают лучам разрушить поражённые болезнью ткани, не подвергая прилегающие к ним участки здоровой кожи даже малейшему риску повреждения.
В некоторых случаях эффективно проведение экстракорпорального фотофореза – облучения крови пациента ультрафиолетовыми лучами. Перед сеансом пациент принимает особый препарат (фотосенсибилизатор), повышающий чувствительность его организма к воздействию света (фото).
При лечении агрессивных форм опухолей химиотерапию совмещают с операцией по трансплантации стволовых кроветворных клеток.
Часто комбинируют различные методы лечения и применяемые препараты. Например, облучение назначается совместно с химиотерапией и после нее.
При своевременном начале лечения и I-II степени злокачественности лимфомы кожи часто удается добиться выраженной ремиссии и продлить жизнь пациента.
Профилактика. Способов, которые могли бы помочь избежать столь серьёзного заболевания, человечество пока не изобрело. Целесообразно по возможности избегать факторов риска, перечисленных ранее. Чтобы, к примеру, не заразиться вирусами гепатита, герпеса или ВИЧ, необходимо пользоваться индивидуальными бритвенными станками и предметами личной гигиены, одноразовыми шприцами (при проведении медицинских манипуляций) и средствами контрацепции барьерного типа при интимной близости.
T-Клеточная лимфома кожи из лимфоцитов CD4
Грибовидный микоз и синдром Сезари представляют собой T-клеточные лимфомы кожи из лимфоцитов CD4 . Примерно 15-20% случаев заканчивается трансформацией в анапластическую крупноклеточную лимфому .
Кожные проявления — бляшки, превращающиеся в опухолевые узлы (грибовидный микоз), или диффузная эксфолиативная эритродермия с появлением опухолевых клеток в крови (синдром Сезари).
Гистологически в эпидермисе и под ним обнаруживается инфильтрация атипичными Т-лимфоцитами с церебриформным ядром ( микроабсцессы Потрие с клетками Лутцнера ). В увеличенных лимфоузлах не всегда обнаруживают опухолевые клетки; в диагностике помогает выявление перестройки генов антигенраспознающих рецепторов Т-лимфоцитов .
Течение. До постановки диагноза больные обычно долго наблюдаются по поводу различных болезней кожи.
Стадии поражения кожи:
Поражение лимфоузлов развивается по мере нарастания поражения кожи. Гистологически подтвержденное поражение лимфоузлов прогностически неблагоприятно.
Поражение внутренних органов. На поздних стадиях могут поражаться любые органы (особенно печень, селезенка, легкие, ЖКТ). Костный мозг затрагивается сравнительно мало, характерна диссеминация по коже и слизистым.
Стадии. Предлагаются разные классификации, в том числе по системе TNM . Вот один из примеров:
— стадия I (лимфоузлы не увеличены); стадия IA — небольшое число бляшек ; стадия IB — большое число бляшек, генерализованное поражение кожи ;
— стадия II (гистологически лимфоузлы не поражены); стадия IIA — бляшки в сочетании с увеличением лимфоузлов ; стадия IIВ — опухолевые инфильтраты в сочетании с увеличением лимфоузлов; стадия III — распространенная эритродермия без гистологических признаков поражения лимфоузлов или внутренних органов;
— стадия IV — гистологические признаки поражения лимфоузлов или внутренних органов.
Прогноз. Около 90% больных на стадии IA переживают 15 лет. При опухолевой стадии или поражении лимфоузлов медиана выживаемости составляет 2-4 года, а при поражении внутренних органов — менее 2 лет.
— Глюкокортикоиды местно часто дают хороший эффект.
— Хлорметин местно используется в инфильтративно-бляшечной стадии. Бывают кожные аллергические реакции.
— Фотохимиотерапия с использованием УФ-А или УФ-В с частотой 3 сеанса в неделю помогает при инфильтративно-бляшечной стадии. Долгосрочный эффект и осложнения изучены плохо.
— Бексаротен представляет собой ретиноид , избирательно связывающийся с ретиноидными RXR-рецепторами . При эритематозной и инфильтративно- бляшечной стадиях препарат эффективен более чем у 60% больных. Бексаротен — единственный ретиноид, разрешенный к применению при этих болезнях.
— Облучение электронным пучком — технически сложный, но эффективный метод лечения, дающий длительные ремиссии (особенно на ранних стадиях болезни).
Химиотерапия и экспериментальные методы терапии приводят лишь к краткосрочному улучшению, не увеличивая выживаемость:
— Химиотерапия рекомендуется только при внекожных поражениях. Монохимиотерапия ( метотрексат , глюкокортикоиды , алкилирующие средства , доксорубицин , гемцитабин , липосомный доксорубицин ) временно помогает 30% больных. Полихимиотерапия (см. CHOP и EPOCH в Приложении Г-2 и Приложении Г-3 ) эффективна у 50% больных и чаще вызывает полную ремиссию.
— Аналоги пуринов — пентостатин , кладрибин и флударабин — помогают примерно 30% больных (флударабин менее эффективен).
— Денилейкин-дифтитокс — химерный белок дифтерийного токсина и ИЛ-2, который применяют при неэффективности других видов лечения. Частое осложнение — снижение иммунитета.
— Моноклональные антитела. При Т-клеточных лимфомах кожи применение моноклональных антител к антигенам Т-лимфоцитов (в том числе конъюгированных с радиоактивными изотопами) приводило к временному улучшению.
— Экстракорпоральная фотохимиотерапия — адъювантная иммунотерапия, являющаяся разновидностью фотохимиотерапии с использованием УФ-А. Лимфоциты больного, полученные с помощью лейкафереза, обрабатываются псораленом и УФ-А и возвращаются больному. Экстракорпоральная фотохимиотерапия индуцирует цитотоксическую иммунную реакцию на идиотипы опухолевых клеток. Она наиболее эффективна в эритематозной стадии болезни.
Лимфома кожи
Лимфома кожи — опухолевые поражения кожи, возникающие в результате злокачественного размножения в ней лимфоцитов. В зависимости от вида размножающихся лимфоцитов различают Т- и В-клеточные лимфомы. Заболевание проявляется образованием на коже узелков, бляшек или эритродермических участков, что сопровождается увеличением лимфатических узлов. Диагностика проводится путем гистологического исследования биопсийного материала из пораженного участка. В лечении лимфомы кожи применяется химиотерапия, лучевая терапия, ПУВА-терапия, экстракорпорального фотофорез.
Лимфома кожи
По данным исследований Т-клеточные лимфомы кожи встречаются в 65-70% случаев, тогда как В-клеточные лимфомы кожи составляют 20-25%. Еще 10% занимают так называемые неклассифицируемые лимфомы кожи.
Причины возникновения лимфомы кожи
Развитие лимфомы кожи связано с мутацией Т или В лимфоцитов, которая приводит к их бесконтрольному размножению и миграции в кожу. Точные причины, запускающие этот механизм не известны. Предполагают, что возникновение злокачественного клона лимфоцитов может быть спровоцировано постоянной антигенной стимуляцией на фоне нарушенной иммунной защиты организма.
Провоцирующую роль отводят вирусным инфекциям, вызванным ретровирусами, цитомегаловирусом, вирусом простого герпеса 8 типа, вирусом Эпштейна—Барра. Действие различных химических веществ и канцерогенов, применяющихся в сельском хозяйстве, химической промышленности, строительстве и других областях, также может быть причиной возникновения лимфомы кожи.
Лимфома кожи бывает первичной, когда заболевание начинается с поражения дермы, и вторичной — в результате миграции лимфоцитов из лимфоидного органа, в котором происходит их размножение. К таким органом относится костный мозг, вилочковая железа, лимфатические узлы, селезенка, лимфоидные скопления по ходу дыхательных путей и желудочно-кишечного тракта.
Симптомы лимфомы кожи
Лимфомы кожи характеризуются полиморфизмом сыпи (пятна, бляшки, узлы), различной степенью выраженности зуда и увеличением периферических лимфатических узлов. По степени злокачественности выделяют лимфомы I, II и III степени. По клиническим проявлениям: узелковую, бляшечную и эритродермическую формы. Узелковая форма Т-клеточной лимфомы кожи I степени характеризуется мелкими плоскими узелками размером с просяное зерно. Узелки имеют сиреневый или желтоватый цвет, располагаются группами и склонны к спонтанному регрессу. При более злокачественном течении узелки увеличиваются, приобретают вишневый цвет и утрачивают тенденцию к группировке. Пациенты погибают от метастазов через 2-5 лет.
Редко встречается мелкоузелковая форма Т-клеточной лимфомы кожи, при которой фолликулярные узелки сливаются в бляшки с напоминающим псориаз поверхностным шелушением. На этом фоне появляются крупные узелки, которые затем подвергаются некрозу. Бляшечная форма Т-клеточной лимфомы кожи I степени представлена нерезко отграниченными бляшками желтоватого цвета. Размер бляшек может быть больше ладони. Они постепенно разрешаются с образованием участков атрофии и гиперпигментации.
Бляшечная форма II степени (грибовидный микоз Алибера) встречается в 26% всех лимфом кожи. Для нее характерно стадийное развитие. Вначале появляются шелушащиеся ярко-розовые пятна и другие элементы (эритематозная стадия). Затем на месте пятен формируются застойно-красные бляшки часто с мокнущей поверхностью и периферическим ростом (бляшечная стадия). В опухолевой стадии бляшки сменяются плоскими узлами размером до апельсина с некрозом в центре образования.
Эритродермическая форма Т-клеточной лимфомы кожи I степени (синдром пре-Сезари) часто развивается на фоне длительно, в течение 10-15 лет, существующей экземы или нейродермита. Кожа покрасневшая и отечная, покрыта крупными пластинками белых чешуек. Наблюдается генерализованное увеличение лимфоузлов, дистрофия ногтей, выпадение волос, лихорадка и мучительный зуд. Через несколько лет пациент может погибнуть от кахексии или процесс переходит в эритродермическую форму II степени (синдром Сезари), характеризующуюся выраженной инфильтрацией, шелушением и сухостью кожи.
Для В-клеточных лимфом кожи характерно отсутствие зуда и других субъективных ощущений при I и II степени злокачественности. Они проявляются бляшечной и узловатой формой. Для бляшечной формы характерны те же стадии, что и для Т-клеточной лимфомы кожи. Узловатая форма развивается с образованием одного или нескольких полушаровидных узлов плотно-эластической консистенции, величина которых достигает размеров грецкого ореха.
Диагностика лимфомы кожи
Во многих случаях лимфомы кожи сопровождаются изменениями в клиническом анализе крови. Для Т-клеточной лимфомы характерны лейкопения и моноцитоз. При синдроме пре-Сезари наблюдается лейкоцитоз и нейтрофилез, повышение количества эозинофилов. При синдроме Сезари может наблюдаться увеличение лейкоцитов до 30000-200000. В-клеточные лимфомы кожи характеризуются возникновением вначале нормохромной, а затем гемолитической анемии.
Решающее диагностическое значение имеет гистологическое и цитологическое исследование материала, взятого путем биопсии элементов лимфомы кожи, а при необходимости и лимфатических узлов. Биопсия позволяет дифференцировать Т и В клеточную лимфому кожи, а также определить степень ее злокачественности. При вовлечении в процесс внутренних органов проводят их исследование: УЗИ брюшной полости, рентгенографию легких, КТ легких и т. п.
Лечение и прогноз лимфомы кожи
Основным методом лечения пациентов с лимфомой кожи является химиотерапия. В ней используют цитостатики (винкристин, винбластин, циклофосфан), кортикостероиды (преднизолон, бетаметазон) и интерфероны (гамма-интерферон). В лечении отдельных пятен, бляшек и единичных опухолей применяют лучевую терапию, ПУВА-терапию, фототерапию. В некоторых случаях эффективно проведение экстракорпорального фотофореза. Часто комбинируют различные методы лечения и применяемые препараты. Например, облучение назначается совместно с химиотерапией и после нее.
При своевременном начале лечения и I-II степени злокачественности лимфомы кожи часто удается добиться выраженной ремиссии и продлить жизнь пациента. К летальному исходу в таком случае приводят интеркуррентные заболевания или осложнения лечения. Если лимфома кожи диагностирована в опухолевой стадии или имеет выраженную злокачественность, прогноз крайне неблагоприятный, летальный исход может наступить через 2 года после начала заболевания.
Дерматология №3 2010 — Т-клеточные лимфомы кожи: современные аспекты лечения
Т-клеточные лимфомы кожи (ТКЛК) – гетерогенная группа лимфопролиферативных заболеваний, обусловленных пролиферацией в коже Т-лимфоцитов с измененным геномом (клональные лимфоциты).
Историческая справка. ТКЛК характеризуются широким разнообразием клинических и гистопатологических проявлений, они объединены в группу экстранодальных злокачественных неходжкинских лимфом с персистированием зрелых лимфоцитов в коже [1, 2]. Среди ТКЛК наиболее изучен грибовидный микоз (ГМ). Изначально термин ГМ появился в связи с тем, что узлы в опухолевой стадии заболевания напоминали шляпки грибов [3]. ГМ составляет приблизительно 1% всех неходжкинских лимфом; средний возраст больных 57 лет с соотношением мужчины/женщины 2:1 [4]. Процент заболевания оценивается в 0,36–0,90 на 100 тыс. человеколет [5]. Данный процент весьма стабилен, хотя общее число случаев заболевания неходжкинскими лимфомами за последние 20 лет увеличилось вдвое и, вероятно, сведения о заболеваемости ГМ занижены [6].
Этиология. Причина ТКЛК неясна. Обсуждается роль в их развитии онкогенных вирусов (таких как вирус Эпштейна–Барр, HTLV-1, HTLV-2, ВПГ-6), также Borrelia burgdorferi [7], профессиональных факторов: химической и физической (ультрафиолетовая, ионизирующая радиации) природы. Вероятна возможность возникновения ТКЛК в ответ на длительное воздействие аллергенов, в том числе лекарственных. На роль иммунных нарушений в развитии ТКЛК указывает преобладание у таких больных Тh2-иммунного ответа с аутокринной стимуляцией интерлейкина (ИЛ)-15, ИЛ-7, ИЛ-2, развитием эозинофилии, увеличением содержания в крови иммуноглобулина E (IgЕ), снижением активности естественных клеток-киллеров [8].
Патогенез. Последние исследования в молекулярной биологии и иммунологии привели к пониманию причин возникновения и циркуляции злокачественных Т-лимфоцитов, аномальной экспрессии цитокинов при росте опухоли, подавления иммунитета, характерного для поздних стадий ГМ, и к пониманию того, что иммунная реакция организма является ключевой для контроля и прогноза болезни. Это позволило сделать существенный прогресс в диагностике и определении стадии заболевания (иммуногистохимический анализ, изучение клональности клеток), а также в терапии (биологическая и целевая терапия) [9].
Патогенез лимфопролиферативных заболеваний – это процесс постепенного накопления генетических дефектов, структурных генов, регулирующих рост, дифференцировку и апоптоз лимфоцитов. Геном атипичного лимфоцита обычно стабилен во времени и содержит несколько хромосомных нарушений. Из-за многочисленных перестроек генов антигенных рецепторов в лимфоцитах часто возникают транслокации с участием локусов Т-клеточного рецептора (ТКР) [10].
Известно, что кожа имеет лимфоидную ткань, ассоциированную с ней и представленную популяцией постоянно рециркулирующих лимфоцитов [8]. Содержание клеток, относимых к иммунной системе кожи, существенно различается в дерме и эпидермисе. Различные популяции и субпопуляции лимфоцитов представлены в дерме в соотношениях, близких к тем, которые определяются в циркулирующей крови. Т-лимфоциты преобладают над В-лимфоцитами, CD4+ Т-клетки (предшественники Т-хелперов) – над CD8+ Т-клетками (будущими цитотоксическими Т-лимфоцитами) [6].
В последние годы тщательно изучаются молекулярно-генетические особенности лимфоцитов. Практически все клетки несут антигенраспознающий рецептор, состоящий из a- и b-цепей, свойственный большинству Т-клеток крови и вторичных лимфоидных органов. Все перечисленные клетки имеют гематогенное происхождение. Из приведенных данных следует, что при миграции из кровяного русла в дерму клетки не подвергаются сколько-нибудь существенной селекции. Миграция клеток в эпидермис строго регламентирована наличием на поверхности клеток молекул адгезии, распознающих определенные молекулярные структуры кератиноцитов, а также рецепторами для хемокинов, секретируемых в эпидермис. В результате в эпидермис проникают лишь избранные разновидности лимфоидных и миелоидных клеток. Так, в эпидермисе определяется необычно высокий процент Т-клеток, имеющий антигенраспознающий рецептор, состоящий из g- и d-цепей ТКР. Известно, что в кожу эти клетки мигрируют из тимуса в эмбриональном периоде, а затем самоподдерживаются за счет медленной пролиферации. Представлены abТ-клетки эпидермиса предшественниками как Т-хелперов (CD4+-клетки), так и цитотоксических Т-лимфоцитов (CD8+ Т-клетки) в сопоставимых количествах. Практически все abТ-клетки относятся к клеткам памяти, т.е. к клеткам, ранее контактировавшим с антигеном [7]. В связи с этим исследования последних лет основаны на проведении молекулярно-генетических анализов с использованием морфологического субстрата биопсийного материала пораженной кожи [11].
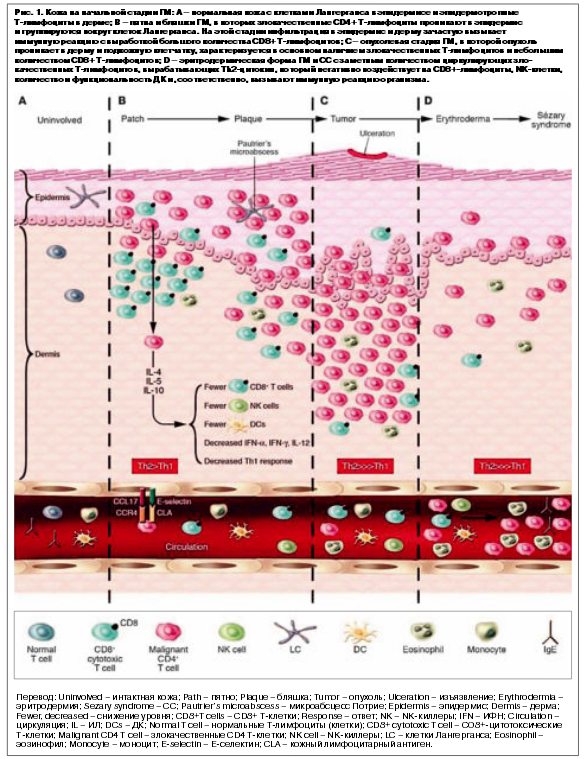
Таким образом, злокачественный клон Т-лимфоцитов при ГМ, вероятно, возникает из нормальных клеток кожи, отвечающих за иммунный ответ. Иммуногистохимические исследования показали, что микроабсцесс Потрие состоит из кластеров злокачественных Т-лимфоцитов, окружающих и находящихся в контакте с клетками Лангерганса, незрелых дендритических клеток (ДК), постоянно находящихся в коже.
Для этих исследований было необходимо установить потенциальную значимость эпидермальных клеток Лангерганса в патогенезе ГМ [12]. Эпидермальные клетки Лангерганса могут быть источником постоянной антигенной стимуляции Т-лимфоцитов, приводящих к активации и впоследствии к клональной экспансии [13] (рис. 1). Таким образом, все опухоли клональны, т.е. происходят из одной клетки, которая претерпела опухолевую трансформацию в результате приобретения нескольких генетических повреждений. Опухолевые клетки более устойчивы к апоптозу, имеют более высокий пролиферативный потенциал по сравнению с нормальными клетками и постепенно начинают преобладать [14]. Установление факта преобладания клональной популяции клеток имеет важное значение в диагностике лимфом кожи, особенно когда дифференциальный диагноз проводится с реактивными состояниями, которые не клональны по определению [15]. Результаты определения клональности, полученные разными методами, могут быть решающими в диагностике, но чаще они дополняют или подтверждают данные других методов исследования.
В 2005 г. WHO-EORTC (Европейская организация по исследованию и лечению рака) выпустила новую классификацию первичных кожных лимфом [16] (см. таблицу).

Рассмотрим часто встречающиеся формы ТКЛК.
ГМ
ГМ – первичная эпидермотропная Т-клеточная лимфома кожи, характеризующаяся пролиферацией малых и средних Т-лимфоцитов с церебриформными ядрами.
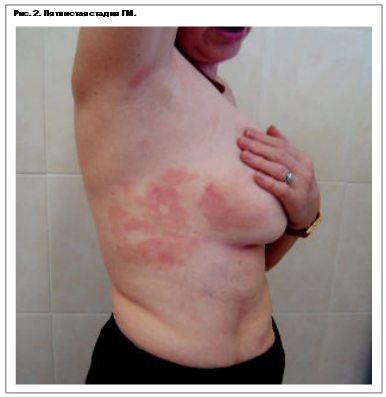
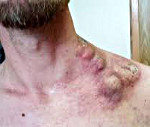
Термин ГМ в настоящее время принято использовать только для классического варианта течения Алибера–Базена, характеризующегося поэтапной эволюцией пятен, бляшек и узлов, или для вариантов со сходным клиническим течением (рис. 2). Заболевание обычно течет благоприятно, медленно прогрессируя в течение нескольких лет или даже 10-летий. На поздних стадиях могут вовлекаться лимфатические узлы и внутренние органы.
Гистологическая картина на ранних стадиях ГМ неспецифична и может симулировать доброкачественный воспалительный дерматоз: периваскулярные инфильтраты в сочетании с псориазиформной гиперплазией эпидермиса, в 1/2 случаев может наблюдаться картина, характерная для гистологической модели поражения зоны дермо-эпидермального соединения [17]. Бляшечная стадия характеризуется плотным полосовидным инфильтратом в верхней части дермы, содержащим высокий процент церебриформных лимфоцитов, и более выраженным эпидермотропизмом. Внутриэпидермальные скопления атипичных лимфоцитов (так называемые микроабсцессы Потрие) являются характерной чертой этой стадии, но встречаются лишь в 10% случаев [18]. С прогрессированием в опухолевую стадию эпидермотропизм исчезает, инфильтрат, состоящий из церебриформных лимфоцитов малых, средних и крупных размеров, бластных клеток и переходных форм в разных пропорциях, становится диффузным и может проникать в подкожную жировую клетчатку.
В поздних стадиях может произойти трансформация ГМ в крупноклеточную лимфому. Критерием трансформации является наличие крупных клеток (с иммунобластной морфологией или крупных плеоморфных клеток), составляющих не менее 25% всех клеток инфильтрата, или их крупные скопления [17]. Большие клетки могут быть CD30+ или CD30-, что не имеет прогностического значения, сама же крупноклеточная трансформация связана с резким ухудшением течения заболевания [19].
Опухолевые клетки при ГМ имеют фенотип зрелых Т-клеток памяти (CD3+, CD45, RO+, CD4+, CD8-), в редких случаях может наблюдаться фенотип CD8+, CD4-. Часто наблюдается демонстрация аберрантного фенотипа (т.е. потеря пан-Т-клеточных антигенов CD2, CD3, CD5, CD7) [13]. Экспрессия цитотоксических протеинов (TIA-1, granzyme B) опухолевыми Т-лимфоцитами может определяться в 10% случаев [20]. В большинстве случаев неопластические лимфоциты обнаруживают клональную перестройку генов, кодирующих a- и/или g-цепь ТКР, специфические хромосомные транслокации пока не были обнаружены [21].
Эритродермия. Иногда первым проявлением ГМ бывает эксфолиативная эритродермия, которая может возникнуть de novo или в результате прогрессии существующих элементов ТКЛК. Кожа при этом диффузно ярко-красная, шелушится, иногда с симметричными островками непораженной кожи. Характерны зуд, повышение температуры тела, слабость, снижение массы тела, лимфаденопатия, алопеция, дистрофия ногтей. Гистологическая картина не отличается от таковой при классическом ГМ [5].
Фолликулотропный ГМ – редкий вариант ГМ, встречающийся, главным образом, у взрослых. Клинически характеризуется интенсивно зудящими, сгруппированными фолликулярными и акнеформными папулами, уплотненными бляшками (в первую очередь в области бровей) и узлами (чаще всего в области головы и шеи), иногда – алопецией (волосистая часть головы, брови) и муцинорреей [3].
Гистологически проявляется перифолликулярным и фолликулярным, нередко неэпидермотропным инфильтратом, состоящим из малых и средних лимфоидных клеток с церебриформными ядрами. Волосяные фолликулы нередко кистозно расширены за счет накопления муцина, с признаками деструкции, процесс может сопровождаться муцинозной дегенерацией фолликулярного эпителия. Фенотипически характеризуется CD4+. Молекулярно-биологически выявляется клональная перестройка генов, кодирующих b- или g-цепь ТКР.
Педжетоидный ретикулез (ПР) – классическая клиническая разновидность с ограниченными проявлениями (тип Woringer-Kolopp) характеризуется развитием четко отграниченных слегка инфильтрированных бляшек округлой, овальной или неправильной формы, цвет которых варьирует от красного до красно-коричневого или красно-фиолетового. Бляшки локализуются на коже дистальных отделов конечностей; поверхность очагов гладкая, блестящая, с незначительным шелушением на отдельных участках, иногда местами гиперкератотическая, бородавчатая. Отличаются медленным периферическим ростом с одновременным разрешением в центре и формированием атрофии и гиперпигментации. Края образованных таким образом очагов кольцевидной или дугообразной конфигурации могут возвышаться, но в отличие от базалиомы не имеют возвышенного валика. Иногда на поверхности бляшки имеется сеточка, напоминающая сетку Уикхема [5].
Соотношение женщин и мужчин 2:1. При разновидности Woringer-Kolopp не поражаются внутренние органы. Заболевание протекает с поражением внутренних органов и часто приводит к летальному исходу.
Гистологическая картина: эпидермис характеризуется гиперкератозом, акантозом. Внутриэпидермально имеется массивный инфильтрат из атипичных лимфоидных клеток с церебриформными ядрами, часто с признаками околоядерного «гало». В дерме могут присутствовать мелкие лимфоидные клетки реактивного характера, макрофаги, эозинофилы обычно отсутствуют.
Фенотип опухолевых клеток: CD3+, CD4+, CD5+, CD8-, описаны случаи с CD8+-фенотипом. Опухолевые клетки также могут экспрессировать СD30.
Молекулярно-генетическое исследование демонстрирует клональную перестройку генов b- или g-цепей ТКР. Течение длительное, с медленным развитием очагов поражения, иногда с их спонтанным регрессом. Таким образом, прогноз для жизни при локализованном варианте относительно благоприятный, хотя возможна диссеминация процесса даже после многолетнего, относительно спокойного локализованного течения процесса. При диссеминации патологического процесса, которая время от времени отмечается у больных с локализованным заболеванием, возможно поражение внутренних органов и летальный исход [14].
СС – форма ТКЛК, при которой практически сразу происходит лейкемизация. Первым проявлением заболевания является эксфолиативная эритродермия с лимфаденопатией и лишь иногда пятна, бляшки или опухоли. Диагноз подтверждается при наличии в периферической крови более 5% атипичных лимфоцитов (клетки Сезари); по иммунофенотипу и генотипу идентичен ГМ, перестройка генов ТКР обнаруживается в опухолевых Т-клетках периферической крови и лимфатических узлов. Гистологически по сравнению с ГМ менее выражены признаки эпидермотропизма; полосовидный лимфоидный инфильтрат расположен преимущественно в дерме. Прогноз плохой. Продолжительность жизни – от 2 до 4 лет. Как и при ГМ, характерный для ранних стадий дерматопатический лимфаденит может смениться специфическим поражением лимфатических узлов. Трансформация в более агрессивную крупноклеточную лимфому может происходить как в коже, клинически проявляясь изъязвленными узлами, так и в лимфатических узлах даже после разрешения эритродермии [5].
Первичные кожные CD30+ Т-клеточные лимфопролиферативные заболевания кожи
Первичные CD30+-лимфопролиферативные заболевания кожи, занимающие 2-е по частоте место в общей структуре ТКЛК (30%), включают первичную анапластическую крупноклеточную лимфому кожи и лимфоматоидный папулез.
1. Лимфоматоидный папулез клинически проявляется высыпаниями (от единичных до нескольких сотен) папулезного и папулонекротического характера в области туловища и проксимальных отделах конечностей, реже – на других участках, склонными к спонтанному регрессу (через 2–8 нед) с исходом в гипо- и гиперпигментированные рубцы. Зуд слабый или отсутствует. Общее состояние не нарушено. В структуре ТКЛК встречается в 18% случаев. Возникает в возрасте от 8 до 60 лет. Соотношение мужчин и женщин примерно одинаковое.
Характеризуется относительно доброкачественным течением, несмотря на гистологическую картину, соответствующую ГМ. Продолжительность ремиссий различна, возможно непрерывное течение. Является лимфомой низкой степени злокачественности [5].
Крупные анапластические клетки обычно имеют фенотип активированных Т-хелперов: СD4+, CD30+, CD25+ и HLADR+, в отдельных случаях СD30-позитивные клетки могут быть CD4-негативны и экспрессировать CD8. Молекулярно-биологически обнаруживается перестройка генов ТКР.
Прогноз заболевания хороший, 5-летняя выживаемость достигает 100%.
2. Первичная анапластическая крупноклеточная Т-лимфома кожи состоит из крупных опухолевых клеток, практически мономорфно экспрессирующих активационный антиген CD30. В отличие от системной анапластической крупноклеточной лимфомы опухолевые клетки реже экспрессируют цитолитические белки, MuM.1 и негативны при реакции с ALK.
При первичной анапластической крупноклеточной лимфоме кожи отсутствуют клинические признаки или анамнез лимфоматоидного папулеза, ГМ или других форм ТКЛК, а доля в структуре ТКЛК оценивается в 12%. Преимущественно поражает лиц мужского пола (2–3:1) в возрасте 60–70 лет.
Клинически заболевание представлено быстро растущим и изъязвленным солитарным узлом (реже – множественными узлами) различной локализации, которые в 10–40% подвергаются спонтанному регрессу. Раньше назывался «грибовидный микоз d`emble» (рис. 3, а). В то же время часты рецидивы. Экстракутанная локализация, при которой преимущественно поражаются регионарные лимфатические узлы, имеет место в 10% случаев.
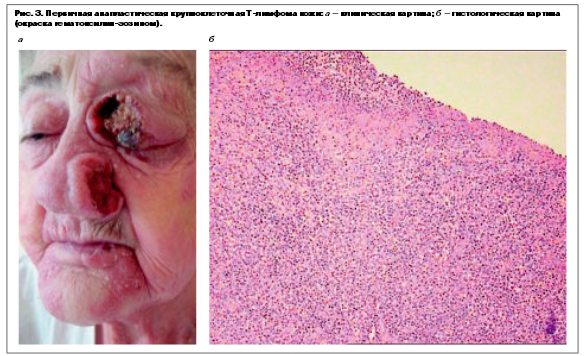
Гистологически проявляется диффузным ростом крупных клеток с полиморфными неправильной формы ядрами, светлой или плотной эозинофильной цитоплазмой, расположенных в дерме, как правило, без признаков эпидермотропизма (рис. 3, б). Активационный антиген CD30+ должен быть экспрессирован более чем в 75–80% опухолевых клеток. Опухолевые клетки экспрессируют CD4+, часто с утратой антигенов CD2, CD5 и/или CD3. Реакция с bF1 как правило негативна в опухолевых клетках, вместе с тем в большинстве случаев можно обнаружить реаранжировку генов ТКР [14].
Прогноз обычно благоприятный, 10-летняя выживаемость достигает 90% случаев.
Диагностика
Полноценная диагностика ТКЛК должна быть комплексной. Для установки диагноза основному количеству пациентов достаточно наличия характерной клинической картины и гистологического метода исследования. При ранней диагностике ТКЛК обязательных клинического и гистологического методов может быть недостаточно. В такой ситуации приходится выбирать между высокой специфичностью и высокой чувствительностью. Иммуногистохимический метод позволяет приблизиться к эталону специфичности. Метод определения клональности с помощью полимеразной цепной реакции на сегодняшний день может считаться более чувствительным [11, 14].
Лечение
При определении методов лечения ТКЛК необходимо учитывать пролиферативный характер процесса, иммунозависимый тип опухолей, обусловленный нарушением взаимодействия иммунокомпетентных структур кожи, наличие воспалительного компонента в очагах поражения, а также эндогенной интоксикации организма, присущей любому опухолевому процессу.
На ранних стадиях развития заболевания при удовлетворительном самочувствии больные ТКЛК не нуждаются в активной противоопухолевой терапии. В этом периоде лечение может быть ограничено применением наружной терапии. Отмечено, что чем позднее начинают применять противоопухолевые цитостатические препараты, тем более длительное время удается сдерживать процесс на стадии «спокойного» клинического течения [15].
Применение наружной терапии (сильнодействующие кортикостероиды – КС, локальные аппликации ретиноидами, ПУВА-терапия или лучевая терапия пучком электронов) в большинстве случаев вызывает усиление апоптоза злокачественных Т-клеток, нередко достаточна для полного регресса высыпаний. При неполном их разрешении добавление одного из системных иммуномодуляторов (интерферон a – ИФН-a или ретиноид бексаротен) обычно приводит к более выраженному эффекту.
Арсенал современных лечебных методов, применяемых в терапии больных ТКЛК, достаточно широк. Это дезинтоксикационные, противовоспалительные и десенсибилизирующие средства, КС и химиотерапевтические препараты, фотохимиотерапевтическое лечение (ПУВА-терапия, фотоферез, фотодинамическая терапия), иммунные препараты (ИФН, интерлейкины – ИЛ, моноклональные антитела, антитимоцитарный глобулин и др.), препараты ретиноевой кислоты, лучевая терапия, наружные средства.
Лечение КС-препаратами (преднизолон, триамцинолон, дексаметазон, метипред) показано почти при всех клинических формах и на разных стадиях развития ТКЛК. Прямым показанием к их назначению является выраженный воспалительный компонент в очагах поражения кожи, чаще всего проявляющийся на начальных стадиях развития ТКЛК и особенно при эритродермических вариантах.
При отсутствии противопоказаний начальная максимальная суточная доза преднизолона может составить от 25 до 60 мг в зависимости от тяжести патологического процесса.
Из клинического опыта можно сделать вывод, что только на начальных стадиях развития ТКЛК можно добиться полной клинической ремиссии с помощью КС-терапии. При бляшечной и опухолевой стадии ГМ, эритродермической форме ГМ и при опухолевых формах ТКЛК лечение КС-препаратами проводится в комплексе с цитостатическими средствами.
Применяют цитостатические препараты разных химических групп: алкилирующие (проспидин, циклофосфамид), антиметаболиты (метотрексат, 6-меркаптопурин), алкалоиды растительного происхождения (винбластин, винкристин), противоопухолевые антибиотики (адриамицин, блеомицин), аналоги пуринов (флюдарабин) и др.
Наиболее эффективным является проспидин, он обладает способностью оказывать цитостатический эффект избирательно на пролиферирующие клетки в коже, т.е. является дерматотропным. Кроме того, оказывает минимальное угнетающее воздействие на кроветворную и иммунную системы организма.
Проспидин вводят внутримышечно ежедневно в суточной дозе 50–200 мг до курсовой дозы 3–4 г, в отдельных случаях до 5–6 г. Через 1,5–2 мес курс лечения повторяют, при необходимости проводят 4–6 курсов. Лечение обычно проводят в сочетании с КС, в частности с преднизолоном, доза которого определяется индивидуально, но обычно находится в пределах 20–40 мг/сут. Клинический эффект чаще всего становится заметным после получения больными 1,5–2 г проспидина. Важно отметить, что препарат обладает выраженным последействием.
В качестве монотерапии может применяться циклофосфан из группы хлорэтиламинов. Препарат вводится внутривенно или внутримышечно в дозе 200–400 мг/сут до 4–6 г на курс, по показаниям можно увеличить до 8–12 г. При клинических вариантах ТКЛК, которые характеризуются высокой степенью злокачественности (опухолевые варианты, СС), проведения монохимиотерапии бывает недостаточно для купирования процесса. Полихимиотерапия обычно включает 2–3 цитостатика, имеющих разный механизм действия:
• Проспидин + винкристин + преднизолон.
• Проспидин + циклофосфан + преднизолон.
• Проспидин + циклофосфан + винкристин + преднизолон.
• Проспидин + метотрексат + преднизолон.
Винкристин (винбластин) вводят 1 раз в неделю внутривенно – соответственно, 0,5–1 и 5–10 мг, всего на курс 5–7 инъекций. В день введения винкристина (винбластина) другие цитостатики не назначаются. Метотрексат при полихимиотерапии назначается по 25 мг внутривенно 1 раз в 5–7 дней до курсовой дозы 150–250 мг. Препарат можно применять перорально по 2,5–5 мг ежедневно до указанной курсовой дозы. В день инъекции метотрексата другие цитостатики не вводятся.
Молекулярно-биологические и иммунологические исследования природы и функций злокачественных Т-клеток, нарушения в экспрессии цитокинов, иммуносупрессия дают основание полагать, что решающую роль в прогрессировании заболевания имеет иммунный ответ больного. Эти данные послужили основанием к новым взглядам на терапию лимфом кожи.
Наиболее активным из биологических препаратов иммунного ответа, используемых в лечении ТКЛК, является природный препарат ИФН-a.
Применение ПУВА-терапии одновременно с ИФН-a дает более высокие результаты в сравнении с монотерапией ПУВА.
Интерфероны. Реаферон по 3 млн Ед внутримышечно ежедневно №10, повторный курс – через 7–10 дней. Виферон 3 млн ЕД ректальные свечи №10.
Индукторы интерферона. Циклоферон по 2 млн ЕД внутримышечно через день №5. Амиксин по 0,125 мг – 1 раз в 2 сут в течение 4 нед. Лейкинферон по 10 тыс. ЕД внутримышечно 2–3 раза в неделю.
Следующим вариантом лечения является фототерапия:
• Фотохимиотерапия (ПУВА).
• Экстракорпоральный фотоферез (ЭКФ).
• Фотодинамическая терапия.
Для больных с циркулирующими злокачественными Т-клетками высоким эффектом обладает ЭКФ, он приводит к массивному апоптозу клеток периферической крови, стимуляции иммунитета, нацеленного на опухоль. Установлена высокая эффективность у пациентов с СС при комбинации ИФН-a, бексаротена и ЭКФ.
У пациентов с тяжелыми проявлениями иммунотерапия с использованием биологических препаратов может привести к клинической ремиссии с эрадикацией злокачественного клона лимфоцитов.
Ретиноиды:
• Бексаротен – от 6–650 мг/м2.
• Изотретиноин 0,5–1 мг/кг в течение 2–3 мес или 0,75 мг/кг в сутки + ИФН 3 млн Ед через день 4 мес.
• Неотигазон 10 мг в день + кальципотриол по 0,5 мг ежедневно 1 мес, после чего доза неотигазона увеличивается до 20 мг.
Современные стратегии терапии лимфом кожи основываются на применении средств, которые условно обозначаются как «биологические препараты». Последние представляют собой рекомбинантные белковые субстанции, полученные биотехнологическим путем из живых клеток животных, растений и микроорганизмов. Механизм действия их основан на специфическом связывании с различными антигенами на мембране опухолевых клеток.
Например, такой препарат, как Дифитокс (ОНТАК). Это белковый конъюгат токсина дифтерии с ИЛ-2, действие которого направлено на Т-клетки, носящие рецептор ИЛ-2 (CD25). После связывания с рецептором ИЛ-2 происходит высвобождение токсина дифтерии, что приводит к блокаде синтеза белка и к апоптозу клеток. Основной механизм действия, как полагают, связан с прямой гибелью злокачественных Т-клеток.
Следует отметить прогресс в подходах к лечению. От парадигмы цитостатической химиотерапии начался переход к современным методам, нацеленным на активацию иммунитета хозяина.